
Leave a Message
We will call you back soon!
 Your message must be between 20-3,000 characters!
Your message must be between 20-3,000 characters!
Please check your E-mail!
SUBMIT

|
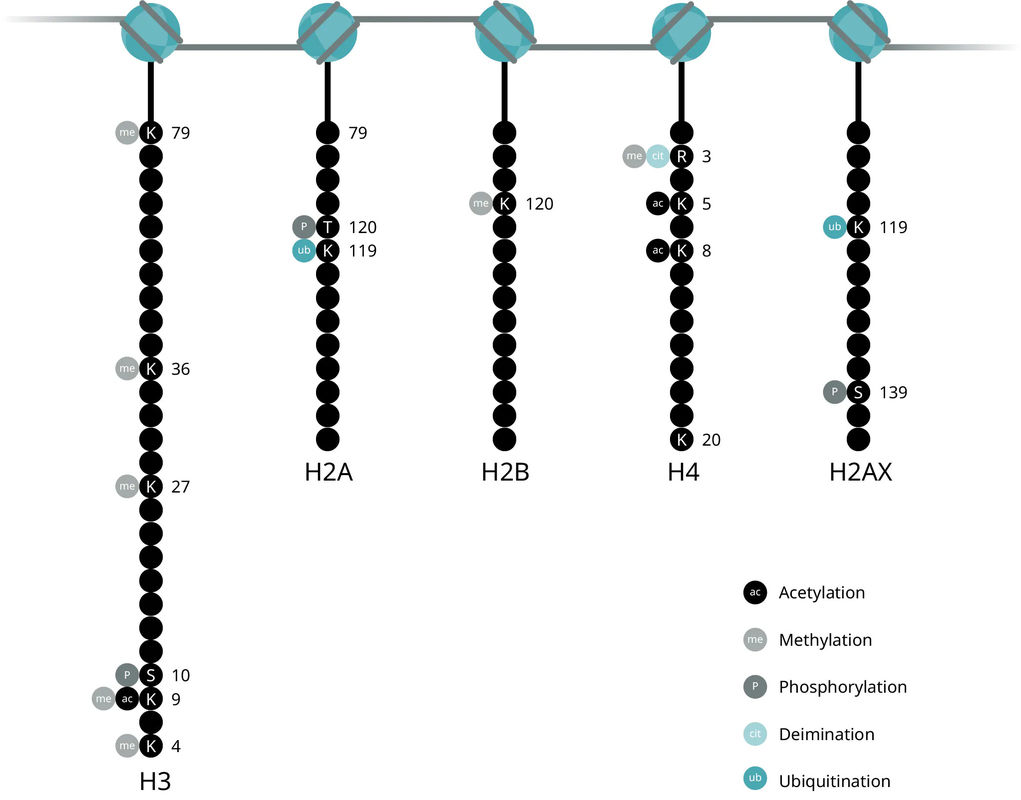
Chromatin architecture, nucleosomal positioning, and ultimately access to DNA for gene transcription, is largely controlled by histone proteins. Each nucleosome is made of two identical subunits, each of which contains four histones: H2A, H2B, H3, and H4. Meanwhile, the H1 protein acts as the linker histone to stabilize internucleosomal DNA and does not form part of the nucleosome itself.
Together, these histone modifications make up what is known as the histone code, which dictates the transcriptional state of the local genomic region. Examining histone modifications at a particular region, or across the genome, can reveal gene activation states, locations of promoters, enhancers, and other gene regulatory elements.
Acetylation is one of the most widely studied histone modifications since it was one of the first discovered to influence transcriptional regulation. Unmodified lysine residues are positively charged but acetylation results in neutralization of the charge on histones, which reduces the interaction of histones and negatively charged DNA. The charge neutralization results in a weaker histone: DNA interaction, allows transcription factor binding and significantly increases gene expression (Roth et al., 2001).
Histone acetylation is involved in cell cycle regulation, cell proliferation, and apoptosis and may play a vital role in regulating many other cellular processes, including cellular differentiation, DNA replication and repair, nuclear import and neuronal repression. An imbalance in the equilibrium of histone acetylation is associated with tumorigenesis and cancer progression.
Acetyl groups are added to lysine residues of histones H3 and H4 by histone acetyltransferases (HAT) and removed by deacetylases (HDAC). Histone acetylation is largely targeted to promoter regions, known as promoter-localized acetylation. For example, acetylation of K9 and K27 on histone H3 (H3K9ac and H3K27ac) is usually associated with enhancers and promoters of active genes. Low levels of global acetylation are also found throughout transcribed genes, whose function remains unclear.
Methylation is added to the lysine or arginine residues of histones H3 and H4, with different impacts on transcription. Arginine methylation promotes transcriptional activation (Greer et al., 2012) while lysine methylation is implicated in both transcriptional activation and repression depending on the methylation site. This flexibility may be explained by the fact that that methylation does not alter histone charge or directly impact histone-DNA interactions, unlike acetylation.
Lysines can be mono-, di-, or tri-methylated, providing further functional diversity to each site of methylation. For example, both mono- and tri-methylation on K4 of histone H3 (H3K4me1and H3K4me3) are activation markers, but with unique nuances: H3K4me1 typically marks transcriptional enhancers, while H3K4me3 marks gene promoters. Meanwhile, tri-methylation of K36 (H3K36me3) is an activation marker associated with transcribed regions in gene bodies.
In contrast, tri-methylation on K9 and K27 of histone H3 (H3K9me3 and H3K27me3) are repressive signals with unique functions: H3K27me3 is a temporary signal at promoter regions that controls development regulators in embryonic stem cells, including Hox and Sox genes. Meanwhile, H3K9me3 is a permanent signal for heterochromatin formation in gene-poor chromosomal regions with tandem repeat structures, such as satellite repeats, telomeres, and pericentromeres. It also marks retrotransposons and specific families of zinc finger genes (KRAB-ZFPs). Both marks are found on the inactive chromosome X, with H3K27me3 at intergenic and silenced coding regions and H3K9me3 predominantly in coding regions of active genes.
Enzymatic regulation
Histone methylation is a stable mark propagated through multiple cell divisions, and for many years was thought to be irreversible. However, it was recently discovered to be an actively regulated and reversible process.
Methylation: histone methyltransferases (HMTs)
SET domain-containing (histone tails)
Non-SET domain-containing (histone cores)
PRMT (protein arginine methyltransferases) family
Demethylation: histone demethylases
KDM1/LSD1 (lysine-specific demethylase 1)
JmjC (Jumonji domain-containing)
PAD4/PADI4
Histone phosphorylation is a critical intermediate step in chromosome condensation during cell division, transcriptional regulation, and DNA damage repair (Rossetto et al., 2012, Kschonsak et al., 2015). Unlike acetylation and methylation, histone phosphorylation establishes interactions between other histone modifications and serves as a platform for effector proteins, which leads to a downstream cascade of events.
Phosphorylation occurs on all core histones, with differential effects on each. Phosphorylation of histone H3 at serine 10 and 28, and histone H2A on T120, are involved in chromatin compaction and the regulation of chromatin structure and function during mitosis. These are important markers of cell cycle and cell growth that are conserved throughout eukaryotes. Phosphorylation of H2AX at S139 (resulting in γH2AX) serves as a recruiting point for DNA damage repair proteins (Lowndes et al., 2005, Pinto et al., 2010) and is one of the earliest events to occur after DNA double-strand breaks. H2B phosphorylation is not as well studies but is found to facilitate apoptosis-related chromatin condensation, DNA fragmentation, and cell death (Füllgrabe et al., 2010).
All histone core proteins can be ubiquitylated, but H2A and H2B are most commonly and are two of the most highly ubiquitylated proteins in the nucleus (Cao et al., 2012). Histone ubiquitylation plays a central role in the DNA damage response.
Monoubiquitylation of histones H2A, H2B, and H2AX is found at sites of DNA double-strand breaks. The most common forms are monoubiquitylated H2A on K119 and H2B on K123 (yeast)/K120 (vertebrates). Monoubiquitylated H2A is also associated with gene silencing, whereas H2B is also associated with transcription activation.
Poly-ubiquitylation is less common but is also important in DNA repair-- polyubiquitylation of H2A and H2AX on K63 provides a recognition site for DNA repair proteins, like RAP80.
Enzymatic regulation
Like other histone modifications, monoubiquitylation of H2A and H2B is reversible and is tightly regulated by histone ubiquitin ligases and deubiquitylating enzymes.
Monoubiquitylation
Polyubiquitylation
Table 1. A cheat sheet for the most common histone modifications and where to find them:
| Histone modification | Function | Location |
| H3K4me1 | Activation | Enhancers |
| H3K4me3 | Activation | Promoters, bivalent domains |
| H3K36me3 | Activation | Gene bodies |
| H3K79me2 | Activation | Gene bodies |
| H3K9Ac | Activation | Enhancers, promoters |
| H3K27Ac | Activation | Enhancers, promoters |
| H4K16Ac | Activation | Repetitive sequences |
| H3K27me3 | Repression | Promoters in gene-rich regions, developmental regulators, bivalent domains |
| H3K9me3 | Repression | Satellite repeats, telomeres, pericentromeres |
| Gamma H2A.X | DNA damage | DNA double-strand breaks |
| H3S10P | DNA replication | Mitotic chromosomes |
ChIP uses antibodies to isolate a protein or modification of interest, along with the DNA to which it is bound (figure 5). The DNA is then sequenced and mapped to the genome to identify the protein or modification’s location and abundance.
![]()
Figure 2: Histone modification ChIP
Antibodies bind directly to modified histone tails. Immunoprecipitation and DNA purification allow for the isolation and identification of the genomic regions that the modifications occupy.
Utilizing antibodies against specific histones and histone modifications in ChIP experiments can reveal the specific locations of
If the function of a histone modification is known, ChIP can identify specific genes and regions with this histone modification signature and the corresponding function across the genome. These genes and regions can then be further examined for their role in the biological process of interest. Using ChIP against H3K4me1, for example, will reveal the locations and sequences of active enhancers throughout the genome, pointing to genes and genetic programs of interest.
Alternatively, if the function of the histone modification is not known, ChIP can identify sequences, genes, and locations with this signature, which can then be used to infer the function of the modification. This technique was pivotal in decoding much of the histone code and is still valuable in ascertaining the function of newly discovered modifications like ubiquitylation and other novel markings.
Histone modifications are dynamically added and removed from histone proteins by specific enzymes (table 2). The balance between these writers and erasers dictates which marks are present on histones, and at what levels, to ultimately control whether specific genetic programs and the cellular processes they orchestrate, are turned on or off.
Table 2. The major categories of histone writers and erasers:
| Modification | Writers | Erasers |
| Acetylation | Histone acetyltransferases (HATs) | Histone deacetylases (HDACs) |
| Methylation | Histone methyltransferases (HMTs/KMTs) and protein arginine methyltransferases (PRMTs) | Lysine demethylases (KDMs) |
| Phosphorylation | Kinases | Phosphatases |
Identifying modification pathways and the specific writers and erasers at play can reveal:
For drug development efforts, compounds can easily be screened for their impact on writer and eraser activity.
In general, histone methyltransferase (HMT) assays are challenging to develop, and most have several drawbacks due to assay design. Typical HMT assays utilize 3H-SAM as a methyl donor and measure S-adenosylhomocysteine (SAH) as a general by-product of the methylation reaction. However, this requires
Abcam HMT activity assays overcome these difficulties, assessing the activity of specific HMTs with antibodies that detect the specific methylated product, providing:
Histone demethylase activity assays typically measure the formation of formaldehyde, a by-product of demethylation. They are therefore susceptible to interference from detergents, thiol groups and a range of ions. Similar to methylation assays, these assays are not specific for any demethylase and can only be performed with purified protein.
Abcam’s histone demethylase assays circumvent these issues by directly measuring the formation of the demethylated product, providing:
Abcam offers kits to analyze overall, as well as H4-specific, HAT activity. These assays measure the HAT-catalyzed transfer of acetyl groups from the Acetyl-CoA donor to histone peptides, which generates the acetylated peptide and CoA-SH. The CoA-SH byproduct is then be measured via colorimetric or fluorometric methods:
HDAC proteins fall into four major groups (class I, class IIA, class IIB, class III, class IV) based on function and DNA sequence similarity. Classes I, IIA, and IIB are considered "classical" HDACs whose activities are inhibited by trichostatin A (TSA), whereas class III is a family of NAD+-dependent proteins (sirtuins (SIRTs)) not affected by TSA. Class IV is considered an atypical class on its own, based solely on DNA sequence similarity to the others.
Each of these classes are associated with different cellular programs and may be assayed individually with various fluorometric assays. For example, SIRTs are typically associated with cancers and neurological diseases. Detecting SIRT activity, or identifying drugs that impact SIRT activity, may point to novel diagnostics or therapeutic strategies for these diseases.
Fluorometric assays utilize an acetylated peptide substrate with a fluorophore and quencher at its amino and carboxyl terminals. Once the substrate is deacetylated, it can be cleaved by a peptidase, releasing the fluorophore from the quencher. The subsequent increase in fluorescence intensity of the fluorophore is directly proportional to deacetylase activity.
It can be useful to inhibit these modifying enzymes using small molecules and then assess downstream consequences to probe the involvement and biological functions of histone modifications. Thus, inhibitors of writers and erasers are vital tools for understanding the roles of epigenetic modification pathways. They are also essential for the validation of “druggable” targets in the context of pre-clinical studies both in academic and industry contexts.
Histone modifications regulate the physical properties of chromatin, and its corresponding transcriptional state, either directly (eg acetyl groups that repel negatively charged DNA to create open chromatin conformation) or via protein adaptors termed effectors. Effector proteins recognize and bind to specific epigenetic marks, and subsequently, recruit molecular machinery to alter chromatin structure. These epigenetic readers determine the functional outcome of histone modifications by translating the histone code into action.
Effector proteins recognize and bind to histone modification marks through effector domains, known as modules (Table 3).
| Histone-binding or effector module | Known histone marks |
| Chromodomain | H3K4me2/3, H3K9me2/3, H3K27me2/3 |
| Tudor | H3K4me3, H4K20me2 |
| MBT | H3K4me1, H4K20me1/2, H1K26me1 |
| WD40 repeats | R2/H3K4me2 |
| Bromodomain | Kac |
| PHD | H3K4, H3K4me3, H3K9me3, K36me3 |
| 41701 | H3S10ph |
| BRCT | H2A.XS139 |
These modules recognize specific histone modifications with amino acids that line the module’s binding pocket. Meanwhile, residues outside of this binding pocket (particularly in the N+2 and N-2 positions) dictate specificity for the histone and amino acid residue being modified (eg H3K4 vs H4K20).
Slight variations in residues within or outside of the binding pocket allow for recognition of similar epigenetic marks. For example, effector proteins can distinguish between mono-, di-, or tri-methylation states with slight variations to the methyl-binding module’s structure. For example, tudor domains may exclusively bind di- or tri-methylated lysines, while PHD finger modules may bind to both, or only to unmodified lysines (Ruthenburg et al., 2007).
Multiple histone-binding modules are often found in the same protein, and/or protein complex, that enable recognition of specific combinations of histone modifications. This allows for a more complex histone code, where histone modifications interact with each other rather than being interpreted in isolation.
Multivalent engagement of histone modifications is important for recognizing discrete marking patterns with composite specificity and enhanced affinity, while also enabling diverse and precise downstream actions. For example, a single epigenetic mark (like H3K4me3) may activate gene transcription in one context, but repress it in another, depending on the surrounding marks. Table 4 shows examples of some of the functional associations of different combinations of histone modifications (Ruthenburg et al., 2007).
Table 4. Functional associations of coexisting histone and DNA modifications:
| Histone marks | Chromatin state |
| H3K4me2/3 + H4K16ac | Transcriptionally active homeotic genes |
| H3K4me2/3 + H3K9/14/18/23ac | Transcriptionally active chromatin |
| H3S10ph + H3K14ac | Mitogen-stimulated transcription |
| H3K4me3 + H3K27me3 | Bivalent domains |
| H3K9me3 + H3K27me3 + 5mC | Silent loci |
| H3K27me3 + H2AK119ub1 | Silent homeotic genes |
| H3K9me3 + H4K20me3 + 5mC | Heterochromatin |
| H3K9me2/3 + H4K20me1+ H4K27me3 + 5mC | Inactive X-chromosome |
Multiple effector modules in a protein or complex may interact with histone modifications on the same, or across, histones and/or nucleosomes. These interactions may be categorized as follows:
Intranucleosomal: binding to the same nucleosome
Internucleosomal: binding to different nucleosomes
Barski, A., Cuddapah, S., Cui, K., Roh, T.Y., Schones, D.E., Wang, Z., Wei, G., Chepelev, I., and Zhao, K. High-resolution profiling of histone methylations in the human genome. Cell 129, 823-837 (2007).
Cao, J. & Yan, Q. Histone ubiquitination and deubiquitination in transcription, DNA damage response, and cancer. Front. Oncol. 2, 26 (2012).
Füllgrabe, J., Hajji, N. & Joseph, B. Cracking the death code: apoptosis-related histone modifications. Cell Death Differ. 17, 1238–1243 (2010).
Greer, E. L. & Shi, Y. Histone methylation: a dynamic mark in health, disease and inheritance. Nat. Rev. Genet. 13, 343–57 (2012).
Kim, J. & Kim, H. Recruitment and biological consequences of histone modification of H3K27me3 and H3K9me3. ILAR J. 53(3-4):232-9 (2012).
Kschonsak, M. & Haering, C. H. Shaping mitotic chromosomes: From classical concepts to molecular mechanisms. BioEssays 755–766 (2015)
Lowndes, N. F. & Toh, G. W.-L. DNA repair: the importance of phosphorylating histone H2AX. Curr. Biol. 15, R99–R102 (2005).
Pinto, D. M. S. & Flaus, A. Structure and function of histone H2AX. Subcell. Biochem. 50, 55–78 (2010).
Rossetto, D., Avvakumov, N. & Côté, J. Histone phosphorylation: A chromatin modification involved in diverse nuclear events. Epigenetics 7, 1098–1108 (2012)
Roth, S. Y., Denu, J. M. & Allis, C. D. Histone acetyltransferases. Annu. Rev. Biochem. 70, 81–120 (2001)
Ruthenburg, A.J., Li, H., Taverna, S.D., Patel, D.J. & Allis, C.D. Multivalent engagement of chromatin modifications by linked binding modules. Nature Rev. Mol. Cell Biol. 8, (2007)
Voigt, P., Tee, W.W., and Reinberg, D. A double take on bivalent promoters. Genes Dev. 27, 1318-1338 (2013).
|
|
Chromatin architecture, nucleosomal positioning, and ultimately access to DNA for gene transcription, is largely controlled by histone proteins. Each nucleosome is made of two identical subunits, each of which contains four histones: H2A, H2B, H3, and H4. Meanwhile, the H1 protein acts as the linker histone to stabilize internucleosomal DNA and does not form part of the nucleosome itself.
Together, these histone modifications make up what is known as the histone code, which dictates the transcriptional state of the local genomic region. Examining histone modifications at a particular region, or across the genome, can reveal gene activation states, locations of promoters, enhancers, and other gene regulatory elements.
Acetylation is one of the most widely studied histone modifications since it was one of the first discovered to influence transcriptional regulation. Unmodified lysine residues are positively charged but acetylation results in neutralization of the charge on histones, which reduces the interaction of histones and negatively charged DNA. The charge neutralization results in a weaker histone: DNA interaction, allows transcription factor binding and significantly increases gene expression (Roth et al., 2001).
Histone acetylation is involved in cell cycle regulation, cell proliferation, and apoptosis and may play a vital role in regulating many other cellular processes, including cellular differentiation, DNA replication and repair, nuclear import and neuronal repression. An imbalance in the equilibrium of histone acetylation is associated with tumorigenesis and cancer progression.
Acetyl groups are added to lysine residues of histones H3 and H4 by histone acetyltransferases (HAT) and removed by deacetylases (HDAC). Histone acetylation is largely targeted to promoter regions, known as promoter-localized acetylation. For example, acetylation of K9 and K27 on histone H3 (H3K9ac and H3K27ac) is usually associated with enhancers and promoters of active genes. Low levels of global acetylation are also found throughout transcribed genes, whose function remains unclear.
Methylation is added to the lysine or arginine residues of histones H3 and H4, with different impacts on transcription. Arginine methylation promotes transcriptional activation (Greer et al., 2012) while lysine methylation is implicated in both transcriptional activation and repression depending on the methylation site. This flexibility may be explained by the fact that that methylation does not alter histone charge or directly impact histone-DNA interactions, unlike acetylation.
Lysines can be mono-, di-, or tri-methylated, providing further functional diversity to each site of methylation. For example, both mono- and tri-methylation on K4 of histone H3 (H3K4me1and H3K4me3) are activation markers, but with unique nuances: H3K4me1 typically marks transcriptional enhancers, while H3K4me3 marks gene promoters. Meanwhile, tri-methylation of K36 (H3K36me3) is an activation marker associated with transcribed regions in gene bodies.
In contrast, tri-methylation on K9 and K27 of histone H3 (H3K9me3 and H3K27me3) are repressive signals with unique functions: H3K27me3 is a temporary signal at promoter regions that controls development regulators in embryonic stem cells, including Hox and Sox genes. Meanwhile, H3K9me3 is a permanent signal for heterochromatin formation in gene-poor chromosomal regions with tandem repeat structures, such as satellite repeats, telomeres, and pericentromeres. It also marks retrotransposons and specific families of zinc finger genes (KRAB-ZFPs). Both marks are found on the inactive chromosome X, with H3K27me3 at intergenic and silenced coding regions and H3K9me3 predominantly in coding regions of active genes.
Enzymatic regulation
Histone methylation is a stable mark propagated through multiple cell divisions, and for many years was thought to be irreversible. However, it was recently discovered to be an actively regulated and reversible process.
Methylation: histone methyltransferases (HMTs)
SET domain-containing (histone tails)
Non-SET domain-containing (histone cores)
PRMT (protein arginine methyltransferases) family
Demethylation: histone demethylases
KDM1/LSD1 (lysine-specific demethylase 1)
JmjC (Jumonji domain-containing)
PAD4/PADI4
Histone phosphorylation is a critical intermediate step in chromosome condensation during cell division, transcriptional regulation, and DNA damage repair (Rossetto et al., 2012, Kschonsak et al., 2015). Unlike acetylation and methylation, histone phosphorylation establishes interactions between other histone modifications and serves as a platform for effector proteins, which leads to a downstream cascade of events.
Phosphorylation occurs on all core histones, with differential effects on each. Phosphorylation of histone H3 at serine 10 and 28, and histone H2A on T120, are involved in chromatin compaction and the regulation of chromatin structure and function during mitosis. These are important markers of cell cycle and cell growth that are conserved throughout eukaryotes. Phosphorylation of H2AX at S139 (resulting in γH2AX) serves as a recruiting point for DNA damage repair proteins (Lowndes et al., 2005, Pinto et al., 2010) and is one of the earliest events to occur after DNA double-strand breaks. H2B phosphorylation is not as well studies but is found to facilitate apoptosis-related chromatin condensation, DNA fragmentation, and cell death (Füllgrabe et al., 2010).
All histone core proteins can be ubiquitylated, but H2A and H2B are most commonly and are two of the most highly ubiquitylated proteins in the nucleus (Cao et al., 2012). Histone ubiquitylation plays a central role in the DNA damage response.
Monoubiquitylation of histones H2A, H2B, and H2AX is found at sites of DNA double-strand breaks. The most common forms are monoubiquitylated H2A on K119 and H2B on K123 (yeast)/K120 (vertebrates). Monoubiquitylated H2A is also associated with gene silencing, whereas H2B is also associated with transcription activation.
Poly-ubiquitylation is less common but is also important in DNA repair-- polyubiquitylation of H2A and H2AX on K63 provides a recognition site for DNA repair proteins, like RAP80.
Enzymatic regulation
Like other histone modifications, monoubiquitylation of H2A and H2B is reversible and is tightly regulated by histone ubiquitin ligases and deubiquitylating enzymes.
Monoubiquitylation
Polyubiquitylation
Table 1. A cheat sheet for the most common histone modifications and where to find them:
| Histone modification | Function | Location |
| H3K4me1 | Activation | Enhancers |
| H3K4me3 | Activation | Promoters, bivalent domains |
| H3K36me3 | Activation | Gene bodies |
| H3K79me2 | Activation | Gene bodies |
| H3K9Ac | Activation | Enhancers, promoters |
| H3K27Ac | Activation | Enhancers, promoters |
| H4K16Ac | Activation | Repetitive sequences |
| H3K27me3 | Repression | Promoters in gene-rich regions, developmental regulators, bivalent domains |
| H3K9me3 | Repression | Satellite repeats, telomeres, pericentromeres |
| Gamma H2A.X | DNA damage | DNA double-strand breaks |
| H3S10P | DNA replication | Mitotic chromosomes |
ChIP uses antibodies to isolate a protein or modification of interest, along with the DNA to which it is bound (figure 5). The DNA is then sequenced and mapped to the genome to identify the protein or modification’s location and abundance.
![]()
Figure 2: Histone modification ChIP
Antibodies bind directly to modified histone tails. Immunoprecipitation and DNA purification allow for the isolation and identification of the genomic regions that the modifications occupy.
Utilizing antibodies against specific histones and histone modifications in ChIP experiments can reveal the specific locations of
If the function of a histone modification is known, ChIP can identify specific genes and regions with this histone modification signature and the corresponding function across the genome. These genes and regions can then be further examined for their role in the biological process of interest. Using ChIP against H3K4me1, for example, will reveal the locations and sequences of active enhancers throughout the genome, pointing to genes and genetic programs of interest.
Alternatively, if the function of the histone modification is not known, ChIP can identify sequences, genes, and locations with this signature, which can then be used to infer the function of the modification. This technique was pivotal in decoding much of the histone code and is still valuable in ascertaining the function of newly discovered modifications like ubiquitylation and other novel markings.
Histone modifications are dynamically added and removed from histone proteins by specific enzymes (table 2). The balance between these writers and erasers dictates which marks are present on histones, and at what levels, to ultimately control whether specific genetic programs and the cellular processes they orchestrate, are turned on or off.
Table 2. The major categories of histone writers and erasers:
| Modification | Writers | Erasers |
| Acetylation | Histone acetyltransferases (HATs) | Histone deacetylases (HDACs) |
| Methylation | Histone methyltransferases (HMTs/KMTs) and protein arginine methyltransferases (PRMTs) | Lysine demethylases (KDMs) |
| Phosphorylation | Kinases | Phosphatases |
Identifying modification pathways and the specific writers and erasers at play can reveal:
For drug development efforts, compounds can easily be screened for their impact on writer and eraser activity.
In general, histone methyltransferase (HMT) assays are challenging to develop, and most have several drawbacks due to assay design. Typical HMT assays utilize 3H-SAM as a methyl donor and measure S-adenosylhomocysteine (SAH) as a general by-product of the methylation reaction. However, this requires
Abcam HMT activity assays overcome these difficulties, assessing the activity of specific HMTs with antibodies that detect the specific methylated product, providing:
Histone demethylase activity assays typically measure the formation of formaldehyde, a by-product of demethylation. They are therefore susceptible to interference from detergents, thiol groups and a range of ions. Similar to methylation assays, these assays are not specific for any demethylase and can only be performed with purified protein.
Abcam’s histone demethylase assays circumvent these issues by directly measuring the formation of the demethylated product, providing:
Abcam offers kits to analyze overall, as well as H4-specific, HAT activity. These assays measure the HAT-catalyzed transfer of acetyl groups from the Acetyl-CoA donor to histone peptides, which generates the acetylated peptide and CoA-SH. The CoA-SH byproduct is then be measured via colorimetric or fluorometric methods:
HDAC proteins fall into four major groups (class I, class IIA, class IIB, class III, class IV) based on function and DNA sequence similarity. Classes I, IIA, and IIB are considered "classical" HDACs whose activities are inhibited by trichostatin A (TSA), whereas class III is a family of NAD+-dependent proteins (sirtuins (SIRTs)) not affected by TSA. Class IV is considered an atypical class on its own, based solely on DNA sequence similarity to the others.
Each of these classes are associated with different cellular programs and may be assayed individually with various fluorometric assays. For example, SIRTs are typically associated with cancers and neurological diseases. Detecting SIRT activity, or identifying drugs that impact SIRT activity, may point to novel diagnostics or therapeutic strategies for these diseases.
Fluorometric assays utilize an acetylated peptide substrate with a fluorophore and quencher at its amino and carboxyl terminals. Once the substrate is deacetylated, it can be cleaved by a peptidase, releasing the fluorophore from the quencher. The subsequent increase in fluorescence intensity of the fluorophore is directly proportional to deacetylase activity.
It can be useful to inhibit these modifying enzymes using small molecules and then assess downstream consequences to probe the involvement and biological functions of histone modifications. Thus, inhibitors of writers and erasers are vital tools for understanding the roles of epigenetic modification pathways. They are also essential for the validation of “druggable” targets in the context of pre-clinical studies both in academic and industry contexts.
Histone modifications regulate the physical properties of chromatin, and its corresponding transcriptional state, either directly (eg acetyl groups that repel negatively charged DNA to create open chromatin conformation) or via protein adaptors termed effectors. Effector proteins recognize and bind to specific epigenetic marks, and subsequently, recruit molecular machinery to alter chromatin structure. These epigenetic readers determine the functional outcome of histone modifications by translating the histone code into action.
Effector proteins recognize and bind to histone modification marks through effector domains, known as modules (Table 3).
| Histone-binding or effector module | Known histone marks |
| Chromodomain | H3K4me2/3, H3K9me2/3, H3K27me2/3 |
| Tudor | H3K4me3, H4K20me2 |
| MBT | H3K4me1, H4K20me1/2, H1K26me1 |
| WD40 repeats | R2/H3K4me2 |
| Bromodomain | Kac |
| PHD | H3K4, H3K4me3, H3K9me3, K36me3 |
| 41701 | H3S10ph |
| BRCT | H2A.XS139 |
These modules recognize specific histone modifications with amino acids that line the module’s binding pocket. Meanwhile, residues outside of this binding pocket (particularly in the N+2 and N-2 positions) dictate specificity for the histone and amino acid residue being modified (eg H3K4 vs H4K20).
Slight variations in residues within or outside of the binding pocket allow for recognition of similar epigenetic marks. For example, effector proteins can distinguish between mono-, di-, or tri-methylation states with slight variations to the methyl-binding module’s structure. For example, tudor domains may exclusively bind di- or tri-methylated lysines, while PHD finger modules may bind to both, or only to unmodified lysines (Ruthenburg et al., 2007).
Multiple histone-binding modules are often found in the same protein, and/or protein complex, that enable recognition of specific combinations of histone modifications. This allows for a more complex histone code, where histone modifications interact with each other rather than being interpreted in isolation.
Multivalent engagement of histone modifications is important for recognizing discrete marking patterns with composite specificity and enhanced affinity, while also enabling diverse and precise downstream actions. For example, a single epigenetic mark (like H3K4me3) may activate gene transcription in one context, but repress it in another, depending on the surrounding marks. Table 4 shows examples of some of the functional associations of different combinations of histone modifications (Ruthenburg et al., 2007).
Table 4. Functional associations of coexisting histone and DNA modifications:
| Histone marks | Chromatin state |
| H3K4me2/3 + H4K16ac | Transcriptionally active homeotic genes |
| H3K4me2/3 + H3K9/14/18/23ac | Transcriptionally active chromatin |
| H3S10ph + H3K14ac | Mitogen-stimulated transcription |
| H3K4me3 + H3K27me3 | Bivalent domains |
| H3K9me3 + H3K27me3 + 5mC | Silent loci |
| H3K27me3 + H2AK119ub1 | Silent homeotic genes |
| H3K9me3 + H4K20me3 + 5mC | Heterochromatin |
| H3K9me2/3 + H4K20me1+ H4K27me3 + 5mC | Inactive X-chromosome |
Multiple effector modules in a protein or complex may interact with histone modifications on the same, or across, histones and/or nucleosomes. These interactions may be categorized as follows:
Intranucleosomal: binding to the same nucleosome
Internucleosomal: binding to different nucleosomes
Barski, A., Cuddapah, S., Cui, K., Roh, T.Y., Schones, D.E., Wang, Z., Wei, G., Chepelev, I., and Zhao, K. High-resolution profiling of histone methylations in the human genome. Cell 129, 823-837 (2007).
Cao, J. & Yan, Q. Histone ubiquitination and deubiquitination in transcription, DNA damage response, and cancer. Front. Oncol. 2, 26 (2012).
Füllgrabe, J., Hajji, N. & Joseph, B. Cracking the death code: apoptosis-related histone modifications. Cell Death Differ. 17, 1238–1243 (2010).
Greer, E. L. & Shi, Y. Histone methylation: a dynamic mark in health, disease and inheritance. Nat. Rev. Genet. 13, 343–57 (2012).
Kim, J. & Kim, H. Recruitment and biological consequences of histone modification of H3K27me3 and H3K9me3. ILAR J. 53(3-4):232-9 (2012).
Kschonsak, M. & Haering, C. H. Shaping mitotic chromosomes: From classical concepts to molecular mechanisms. BioEssays 755–766 (2015)
Lowndes, N. F. & Toh, G. W.-L. DNA repair: the importance of phosphorylating histone H2AX. Curr. Biol. 15, R99–R102 (2005).
Pinto, D. M. S. & Flaus, A. Structure and function of histone H2AX. Subcell. Biochem. 50, 55–78 (2010).
Rossetto, D., Avvakumov, N. & Côté, J. Histone phosphorylation: A chromatin modification involved in diverse nuclear events. Epigenetics 7, 1098–1108 (2012)
Roth, S. Y., Denu, J. M. & Allis, C. D. Histone acetyltransferases. Annu. Rev. Biochem. 70, 81–120 (2001)
Ruthenburg, A.J., Li, H., Taverna, S.D., Patel, D.J. & Allis, C.D. Multivalent engagement of chromatin modifications by linked binding modules. Nature Rev. Mol. Cell Biol. 8, (2007)
Voigt, P., Tee, W.W., and Reinberg, D. A double take on bivalent promoters. Genes Dev. 27, 1318-1338 (2013).